News
Guidelines for Molecular Counting Using Fluorescent Proteins
Guidelines for Molecular Counting Using Fluorescent Proteins
![]() By eHealth Network - 16 January 2014
By eHealth Network - 16 January 2014


To know how many proteins assemble together at the nanoscale is fundamental for understanding protein function. Sometimes, proteins must be in an “oligomeric” state to be functional, although “oligomerization” of certain proteins can also lead to diseases. The ability to determine protein stoichiometry and monitor changes in the balance between monomeric, dimeric and multi-meric proteins can allow scientists to see the differences between a properly functioning cell and a diseased cell. Therefore, there is a great interest in being able to count proteins and determine their stoichiometry.
In a recent study carried out at ICFO, the Institute of Photonic Sciences, the research group of Advanced fluorescence imaging and biophysics, led by Nest Fellow Dr. Melike Lakadamyali was able to quantify the photoactivation efficiency of all the known “ir-reversibly photoswitching fluorescent proteins” and establish a proper detailed reference framework for determining protein stoichiometry. To do this, they used a nanotemplate of known stoichiometry (the human Glycine receptor expressed in Xenopus oocytes) and studied several fluorescent proteins to see the percentage of proteins that was photoactivated. The results of this study have recently been published in Nature Methods.
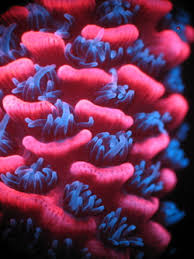
Human Glycine Receptor
Human glycine receptor, which has a well-defined subunit stoichiometry of two “beta” and three “alpha”-subunits was used as a nanotemplate by attaching fluorescent proteins to these subunits to measure photoactivation efficiency at the single molecule level.
Image credit: ICFO
“Molecular counting” is becoming a closer reality thanks to the discovery of photoactivatable fluorescent proteins and the development of super resolution microscopy. Photoactivatable flourescent proteins change their fluorescence property from dark to bright when exposed to light (i.e. laser light). Through the use of localization-based super-resolution microscopy, researchers are able to photoactivate, image and follow these genetically encoded fluorescent proteins, one at a time, to study what is happening inside a cell at the molecular level. However, despite the “molecular counting” ability that seems intrinsic to the imaging strategy (activating one fluorescent protein at a time should also allow counting how many total fluorescent proteins exist) relating the number of counted fluorescent proteins to actual protein stoichiometry has been difficult. One important reason for this difficult task in counting has been the fact that, until recently, it was not known whether all fluorescent proteins become bright when exposed to laser light. Failure to photoactivate would lead to undercounting since a fraction of probes would be dark and never appear in the image.

By measuring the photoactivation efficiency as well as other photophysical factors such as blinking, the group determined which proteins are best suited for molecular counting and found that none are perfect (most fluorescent proteins fail to photoactivate about 50% of the time). Therefore, they emphasized the importance of taking into account the photoactivation efficiency to correctly interpret the quantitative information.

Dr. Lakadamyali states that “at the moment we have to work with imperfect fluorescent proteins and take into account the limitations when quantifying protein stoichiometry, however, now that there is a robust way to characterize photoactivation efficiency, future fluorescent protein engineering work will likely focus on optimizing this parameter for generating fluorescent proteins better suited for molecular counting using super-resolution”.
The group combines several cutting-edge single molecule imaging techniques to study how protein organization, dynamics and stoichiometry relate to protein function in several fundamental biological processes, such as intracellular transport, autoimmune neurological disorders or stem cell reprogramming

 Like
Like
 Dislike
Dislike
Be a part of Elets Collaborative Initiatives. Join Us for Upcoming Events and explore business opportunities. Like us on Facebook , connect with us on LinkedIn and follow us on Twitter , Instagram.
"Exciting news! Elets technomedia is now on WhatsApp Channels Subscribe today by clicking the link and stay updated with the latest insights!" Click here!